SHORT REPORT
Doi: 10.5578/tt.22093
Tuberk Toraks 2017;65(1):56-59

İmm?nyetmezliğin arkasına gizlenen t?berk?loz;
kronik gran?lomat?z hastalık tanısı alan iki olgunun irdelenmesi
Hikmet Tekin NACAROĞLU1, Semiha BAH?ECİ ERDEM1, Nesrin G?LEZ1, Canan Şule ?NSAL KARKINER1,
İlker DEVRİM2, Ferah GENEL1, Mustafa Yavuz K?KER3, Demet CAN1
1 Clinic of Pediatric Immunology and Allergy, Dr. Beh?e Uz Children Diseases Training and Research Hospital, Izmir, Turkey
1 Dr. Beh?et Uz ?ocuk Hastalıkları Eğitim ve Araştırma Hastanesi, ?ocuk İmm?noloji ve Allerji Kliniği, İzmir, T?rkiye
2 Clinic of Pediatric Infectious Diseases, Dr. Beh?et Uz Children Diseases Training and Research Hospital, Izmir, Turkey
2 Dr. Beh?et Uz ?ocuk Hastalıkları Eğitim ve Araştırma Hastanesi, ?ocuk İnfeksiyon Hastalıkları Kliniği, İzmir, T?rkiye
3 Department of Pediatric Immundology and Allergy, Faculty of Medicine, Erciyes University, Kayseri, Turkey
3 Erciyes ?niversitesi Tıp Fak?ltesi, ?ocuk İmm?noloji ve Allerji Bilim Dalı, Kayseri, T?rkiye
?ZET
İmm?nyetmezliğin arkasına gizlenen t?berk?loz; kronik gran?lomat?z hastalık tanısı alan iki olgunun irdelenmesi
Kronik gran?lomat?z hastalık (KGH), nikotinamid adenin din?kleotit fosfat (NADPH) oksidaz sistemindeki defektlere bağlı olarak gelişen, tekrarlayan ve yaşamı tehdit eden infeksiyonlar ve artmış inflamatuvar yanıta bağlı gran?lom oluşumu ile karakterize heterojen, kalıtsal primer bir imm?nyetmezlik hastalığıdır. Akciğer, deri, lenf nodları ve karaciğer infeksiyon nedeni ile en ?ok tutulan organlardır. Akciğer tutulumuna bağlı olarak yineleyen pn?moni, hiler lenfadenopati, ampiyem, apse, retik?lonod?ler g?r?nt?ler ve gran?lomalarla g?r?lebilir. Son yıllarda da KGH olgularında mikobakteriyel hastalıklara yatkınlık olduğu bildirilmektedir. Bu makalede uzamış pn?moni nedeniyle tetkik edilirken KGH ve t?berk?loz tanısı alan biri 18 aylık diğeri 5 yaşında olan iki erkek olgu sunulmuştur. Bu bildiri KGH'nin sadece deri apseleri, Aspergillus infeksiyonu varlığında değil, BCG-itis ve/veya t?berk?lozlu olgularda da ayırıcı tanıda akla gelmesi ve mikobakteri infeksiyonlarının hastalığın seyrinde g?r?lebileceğinin akılda tutulması gerektiği i?in sunulmuştur.
Anahtar kelimeler: Kronik gran?lamat?z hastalık, imm?nyetmezlik, fagositer bozukluk, mikobakteriyel hastalıklar
SUMMARY
Tuberculosis masked by immunodeficiency: a review of two cases diagnosed with chronic granulomatous disease
Chronic granulomatous disease (CGD) is a genetically heterogeneous primary immunodeficiency that is characterized by recurrent and life-threatening infections resulting from defects in phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system and granuloma formation due to increased inflammatory response. The most commonly involved organs are the lungs, skin, lymph nodes, and liver due to infection. It may present with recurrent pneumonia, hilar lymphadenopathy, empyema, abscess, reticulonodular patterns, and granulomas due to lung involvement. In recent years, mycobacterial disease susceptibility has been reported in CGD cases. This article presents two male cases, one of whom is aged 18 months and the other is aged 5 years, who were diagnosed with CGD and tuberculosis during examination due to extended pneumonia. This report is presented because CGD should be considered not only in the presence of skin abscesses and Aspergillus infections, but also in the differential diagnosis for cases with BCG-itis and/or tuberculosis. It should be kept in mind that mycobacterial infections can occur during the course of the disease.
Key words: Chronic granulomatous disease, immunodeficiency, phagocytic disorder, mycobacterial disease
Geliş Tarihi/Received: 23.02.2016 • Kabul Ediliş Tarihi/Accepted: 19.06.2016
INTRODUCTION
Chronic granulomatous disease (CGD) is a rare genetic disease and a disorder of phagocyte function that is characterized by recurrent infections by catalase-positive bacteria and fungi (1). It is a heterogeneous disease and has a wide range of clinical findings and prognosis due to different genotypic and phenotypic interactions; however, a common feature of CGD cases is the recurrent severe bacterial and fungal infections from the infantile period or childhood. In recent years, mycobacterial disease susceptibility has been also reported in CGD cases (2-5). It is difficult to diagnose because of the challenges of demonstrating bacilli in diagnosing mycobacterial infections in the clinical practice. This becomes even more difficult, especially in cases with immunodeficiency. This article presents two cases diagnosed with CGD and tuberculosis during examination due to extended pneumonia.
Cases
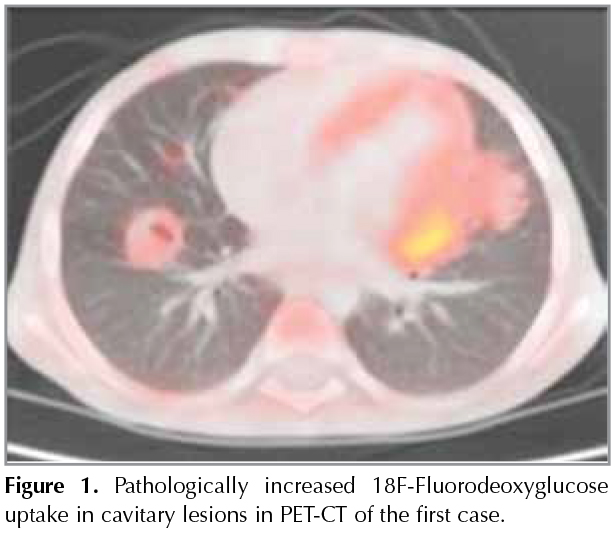
First Case: A 5-year-old male patient presented at an external center with fever, cough, abdominal pain, and night sweats, and was monitored in the hospital for ten days with the diagnosis of pneumonia, and referred to our hospital due to no clinical or radiological improvement. His personal history involved treatment due to axillary lymphadenitis at the age of three. In his physical examination, body weight was 14.9 kg (3-10p) and height was 104 cm (10-25p). His respiratory system examination revealed decreased breath sounds bilaterally at the bases. The patient had hepatosplenomegaly and there was no particular finding in other system examinations. Laboratory tests revealed the following: Hb= 10.4 g/dL, WBC= 15.600/mm3, platelets= 631.000/mm3, C-reactive protein 4.22 mg/dL, and erytrhocyte sedimentation rate (ESR)= 35 mm/h; blood biochemical tests were normal. Thoracic CT imaging of the patient revealed bilateral hilar lymphadenopathy and parenchymal nodular densities. In the tests for etiology, the tuberculin skin test (TST) was non-reactive, QuantiFERON was negative; and in fasting gastric juice, acid-alcohol resistant bacilli (AARB) was (-) and PCR was (-); there was no growth in the mycobacterial culture. The tested immunoglobulin levels revealed hypergammaglobulinemia (IgG= 1800 mg/dL, IgM= 112 mg/dL, IgA= 301 mg/dL), and IgE was 38.66 IU/mL; the levels of C3 and C4 were normal for his age. The diagnostic bronchoscopy revealed normal lower airway anatomy. In the bronchoalveolar lavage sample examination; AARB was (-), PCR was (-), and there was no growth in mycobacterial culture. The positron emission tomography-computed tomography that was performed for malignancy revealed pathologically increased 18F-fluorodeoxyglucose (FDG) uptakes in the cavitary lesion (Figure 1). The clinical evaluation and histopathological verification of the findings were recommended for malignancy or granulomatous processes. For a pathological diagnosis with the available findings, a biopsy was performed from the pulmonary lesion, guided by video-assisted thoracoscopic surgery (VATS). The pathology was concluded as caseified granulomatous inflammation and the patient was initiated with anti-tuberculosis treatment. In the control thoracic CT examination at month two of the treatment, the nodular lesions were improved with atelectasis and the hilar lymphadenopathies were diminished. The myeloperoxidase activity was 100%; the phagoburst activity was lower as 56.5% and 62.3%. Therefore, the diagnosis of autosomal recessive CGD was considered for the patient. The genetic analysis revealed P22 phox defect (AR-CGD, hom. c.416G > A; p.Arg139Gln). The patient was initiated with trimethoprim/sulfamethoxazole and itraconazole prophylaxis and he is continuing tuberculosis treatment and under follow-up.
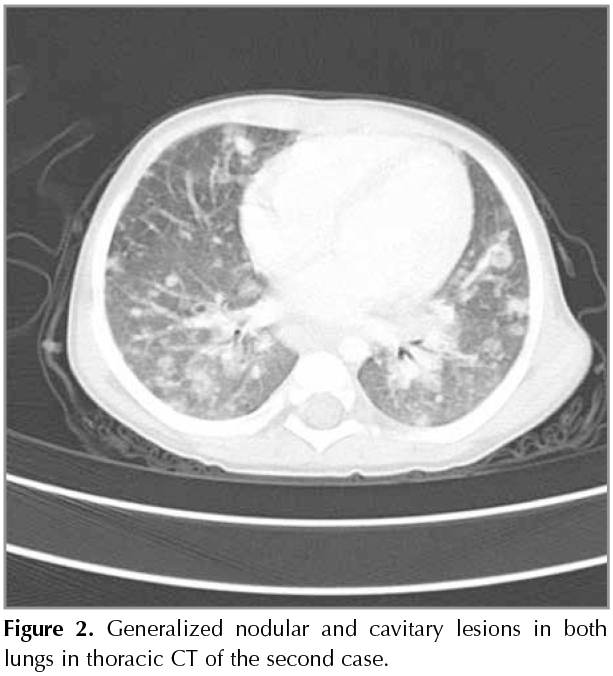
Second Case: An 18-month-old male patient had fever for the last two months and was admitted with the diagnosis of a fever of unknown origin. His personal history involved hospitalization due to thrombocytopenia etiology when he was 27 days old, frequent infections, and repeated hospitalizations due to hepatosplenomegaly, and there was no particular findings in his history. In his physical examination, body weight was 10 kg (10p), height was 76 cm (3p), vital findings were stable, and there were no pathological findings other than hepatosplenomegaly in the system examination. The laboratory tests revealed the following: Hb= 8.5 g/dL, WBC= 20.800 mm3, platelets= 440.000 mm3, ESR= 57 mm/h, and C-reactive protein= 12.4 g/dL; blood biochemical parameters were normal. Chest radiography showed an infiltrative appearance and the thoracic CT revealed generalized nodular/cavitary lesions and hilar lymphadenopathies in both lungs (Figure 2). The tested immunoglobulin levels revealed hypergammaglobulinemia (IgG= 1349, IgM= 236, and IgA= 233), and the levels of C3 and C4 were normal for his age. The lymphocyte subgroup distribution was normal. Oxidative burst activity was not found in the neutrophils and monocytes, and the patient was considered consistent with chronic granulomatous disease with clinical and laboratory findings. The genetic analysis revealed gp91phox defect (X-CGD). The patient was not a tuberculosis index case, and TST was 15 x 20 mm. In the fasting gastric juice: ARB was (-), PCR was (-), and there was no growth in the mycobacterial culture. After the QuantiFERON test was also found positive, the patient was evaluated with clinical, radiological, and laboratory findings, and diagnosed with tuberculosis. He was initiated with anti-tuberculosis treatment, as well as interferon gamma therapy (50 ?g/m2; SC; three days a week) for CGD. There was an improvement in the thoracic CT performed at month two of treatment. The patient is still under follow-up and being examined for bone marrow transplantation.
Discussion
CGD is a genetically heterogeneous primary immunodeficiency that is characterized by recurrent and life-threatening infections resulting from defects in phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system and granuloma formation due to increased inflammatory response. The frequency of the disease is 1:200.000-250.000/live births in the United States (1-4). CGD can occur at any age from early childhood to adulthood; however, it is usually diagnosed through recurrent infections and granuloma formation in the early childhood or at the age of five. In general, X-linked CGD is diagnosed at an earlier age than p47phox and severe infections occurs at an earlier age (3,4). The two cases presented here were also aged five and below, the first case had P22 phox defect (AR-CGD) and the second case had gp91phox defect (X-CGD) mutation.
In recent years, mycobacterial disease susceptibility has been reported in CGD cases. The mycobacterial infection susceptibility in CGD is indicated to result from the reduced clearance of mycobacteria, which are catalase-positive organisms, and prolonged inflammatory activity caused by the impaired oxidative burst activity in macrophages (4). The incidence of pneumonia due to mycobacterial infections is reported to be 6% in records from the United States, while this rate is reported to be much higher in countries where tuberculosis is endemic (5-8). Bustamante et al. reviewed literature for the coexistence of mycobacterial disease and CGD, and reported that they found BCG-itis in 38 patients, atypical mycobacterial infection in four patients, tuberculosis in 16 patients, tuberculosis and BCG-itis in seven patients, and mycobacterial infection by Mycobacterium spp. in seven patients (6). A study by Ying et al. reported 52.08% as the incidence of mycobacteria as a pathogen microorganism in 48 CGD patients and established local skin infection after BCG vaccine in 22 cases (7). Another study from China also reported pulmonary tuberculosis due to Mycobacterium tuberculosis in 7 of 17 patients diagnosed with X-linked CGD, and that seven patients had prolonged scarring or abscess formation at the BCG injection site, and one had disseminated BCG infection (8). A European study, which evaluated 429 CGD cases, established local skin infection after BCG vaccine in 24 cases, and reported that BCG was isolated from the lymph nodes in ten cases and the incidence of BCG-itis was 8% (9).
CGD patients are followed-up with long-term prophylactic low-dose antibiotic and antifungal therapy. Antimicrobial prophylaxis consists of the triad of trimethoprim/sulfamethoxazole, itraconazole, and IFN-, an immunostimulant therapy. The IFN- therapy administered to patients with chronic granulomatous disease is included in treatment protocols since a partial improvement was found in vitro in superoxide production in the monocytes and granulocytes, and the frequency of infection, the frequency of hospitalization, and the quality of life are improved without any significant toxicity. IFN- is the key cytokine in mycobacterial immunity, and therefore, there is a high indication for using IFN- in regions endemic for tuberculosis (10). Both our cases presented here, IFN- therapy were initiated and stem cell transplantations were planned.
CGD should be considered not only in the presence of skin abscesses and Aspergillus infection, but also in the differential diagnosis for the cases with BCG-itis and/or tuberculosis. It should be kept in mind that mycobacterial infections can occur during the course of this disease. Especially if there is a clinical appearance inconsistent with the severity of chest radiography, immunodeficiency presentations should be considered such as CGD; it should kept in mind that tuberculosis, which does not manifest its classic presentation due to immunodeficiency, may accompany the disease.
REFERENCES
- K?ker MY, Camcıoğlu Y, van Leeuwen K, Kılı? SŞ, Barlan I, Yılmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol 2013;132:1156-63.
- Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine 2000;79:170-200.
- Gill HK, Kumar HC, Cheng CK, Ming CC, Nallusamy R, Yusoff NM, et al. X-linked chronic granulomatous disease in a male child with an X-CGD carrier, Klinefelter brother. Asian Pac J Allergy Immunol 2013;31:167-72.
- Kusuhara K, Ohga S, Hoshina T, Saito M, Sasaki Y, Ishimura M, et al. Disseminated Bacillus Calmette-Gu?rin lymphadenitis in a patient with gp91phox- chronic granulomatous disease 25 years after vaccination. Eur J Pediatr 2009;168: 745-7.
- Lau YL, Chan GC, Ha SY, Hui YF, Yuen KY. The role of phagocytic respiratory burst in host defense against Mycobacterium tuberculosis. Clin Infect Dis 1998;26:226-7.
- Bustamante J, Aksu G, Vogt G, de Beaucoudrey L, Genel F, Chapgier A, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol 2007;120:32-8.
- Ying WJ, Wang XC, Sun JQ, Liu DR, Yu YH, Wang JY. Clinical features of chronic granulomatous disease. Zhonghua Er Ke Za Zhi 2012;50:380-5.
- Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J 2008;27:224-30.
- Van den Berg JM, van Koppen E, Belohradsky BH, Bernatowska E, Corbeel L, Espanol T, et al. Chronic granulomatous disease: the European experience. PLoS One 2009;4:e5234.
- North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol 2004;22:599-623.
Yazışma Adresi (Address for Correspondence)
Dr. Hikmet Tekin NACAROĞLU
Dr. Beh?et Uz ?ocuk Hastalıkları Eğitim ve
Araştırma Hastanesi, ?ocuk İmm?noloji ve Allerji Kliniği,
İZMİR - TURKEY
e-mail: tekin212@gmail.com