CASE REPORT
Doi: 10.5578/tt.8900
Tuberk Toraks 2016;64(1):73-76

?oklu pulmoner nod?l?n nadir sebebi; herediter hemorajik telenjiektazi
Şule KOŞAR1, Bet?l KIZILDAĞ2, Arzu CANAN3, Ozan KARATAĞ1, Uğur G?NL?G?R4, Abdullah SARIYILDIRIM1
1 Onsekiz Mart ?niversitesi Tıp Fak?ltesi, Radyoloji Anabilim Dalı, ?anakkale, T?rkiye
1 Department of Radiology, Faculty of Medicine, Onsekiz Mart University, Canakkale, Turkey
2 Kahramanmaraş S?t?? İmam ?niversitesi Tıp Fak?ltesi, Radyoloji Anabilim Dalı, Kahramanmaraş, T?rkiye
2 Department of Radiology, Faculty of Medicine, Kahramanmaras Sutcu Imam University, Kahramanmaras, Turkey
3 Antalya Atat?rk Devlet Hastanesi, Radyoloji Kliniği, Antalya, T?rkiye
3 Clinic of Radiology, Antalya Ataturk State Hospital, Antalya, Turkey
4 Onsekiz Mart ?niversitesi Tıp Fak?ltesi, G?ğ?s Hastalıkları Anabilim Dalı, ?anakkale, T?rkiye
4 Department of Chest Diseases, Faculty of Medicine, Onsekiz Mart University, Canakkale, Turkey
?ZET
?oklu pulmoner nod?l?n nadir sebebi; herediter hemorajik telenjiektazi
Herediter hemorajik telenjiektazi diğer adıyla Rendu-Osler-Weber sendromu nadir g?r?len herediter bir hastalıktır. Tanısında rek?rren epistaksis, telenjiektazi, visseral arteriyoven?z malformasyonların (AVM) ve aile ?yk?s?n?n varlığı ?nem taşımaktadır. AVM'ler karaciğer, beyin ya da akciğerde bulunabilir. ve kitleleri taklit edebilir. Radyolojik değerlendirme hastalığın tanısında ve tedavinin y?nlendirilmesinde b?y?k ?nem taşımaktadır. Bu nedenle, ?zellikle AVM saptanan hastalarda epistaksis ve aile ?yk?s?n?n varlğında radyologlar HHT tanısını akılda bulundurmalıdırlar. Bu yazıda, malignite ş?phesiyle girişimsel radyolojiye biyopsi amacıyla g?nderilen ve HHT'ye ikincil pulmoner AVM saptanan bir hastayı sunduk.
Anahtar kelimeler: Herediter hemorajik telenjiektazi, pulmoner arteriyoven?z malformasyon, bilgisayarlı tomografi
SUMMARY
An uncommon cause of multiple pulmonary nodules; hereditary hemorrhagic telangiectasia
Hereditary hemorrhagic telangiectasia (HHT), or Rendu-Osler-Weber syndrome (ROWS) is a very rare hereditary disease. The diagnosis is based on the clinical findings such as recurrent epistaxis, telangiectases, visceral arteriovenous malformations (AVMs) and family history. AVMs are found in the liver, lung or brain and could mimick the masses of these organs. Radiologic evaluation plays a critical role during diagnostic and therapeutic management of ROWS. Hence, radiologists should be aware of the diagnosis of HHT in the patients with AVMs, history of epistaxis and family history. We report a patient with multiple pulmonary AVMs secondary to HHT who has referred to our interventional radiology department for computed tomography guided transthorasic lung biopsy procedure with suspicious of malignancy.
Key words: Hereditary hemorrhagic telangiectasia, pulmonary arteriovenous malformations, computed tomography
Geliş Tarihi/Received: 07.01.2015 • Kabul Ediliş Tarihi/Accepted: 21.01.2015
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Rendu-Osler-Weber syndrome (ROWS), is a rare vascular dysplasia with an autosomal dominant inheritance habit. The prevalence of HHT is estimated approximately 1-2/100.000. The clinical diagnosis of HHT includes spontaneous and recurrent epistaxis, telangiectases, visceral arteriovenous malformations (AVMs) and family history (1). Besides the clinical presentation; state of art in computed tomography (CT) and magnetic resonance imaging (MRI) techniques, not only demonstrate the involvement of vascular structures or organs also provide the manifestation of the whole spectrum of the disease as well.
We report a patient with pulmonary AVMs and hepatic telangiectases, secondary to HHT who had initially referred to our department for biopsy procedure with suspicious of malignancy.
Case report
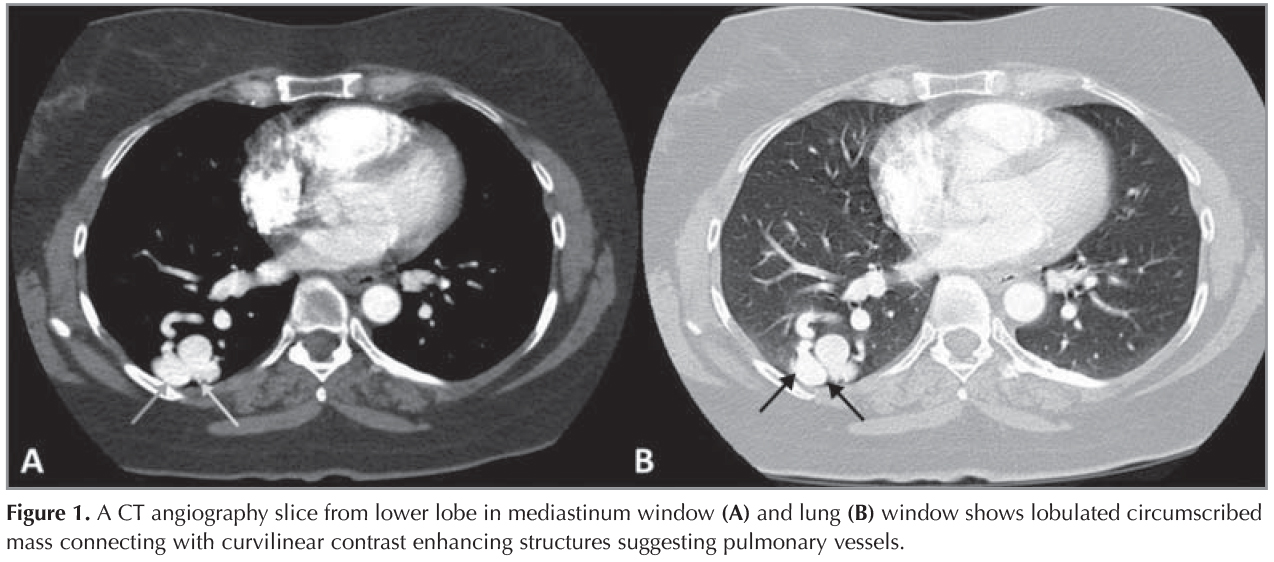
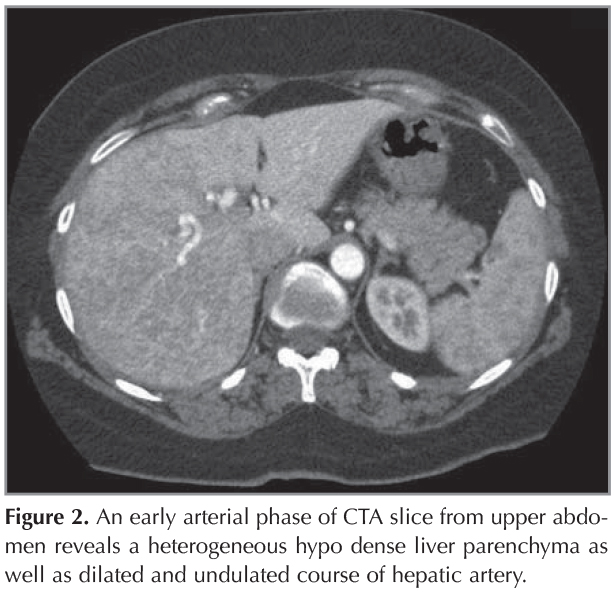
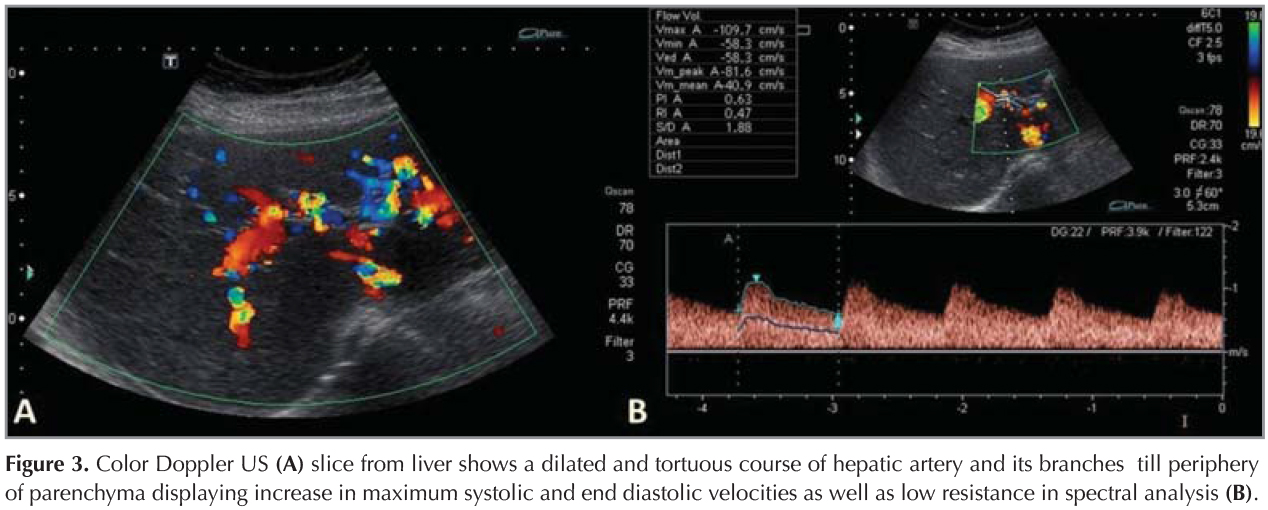
A 44 year-old female patient who had dyspnea aggravating with exercise for last three months was admitted the state hospital. Unenhanced chest CT which had achieved suspicious of asthma, revealed multiple pulmonary nodules in both lungs. CT-guided cutting needle biopsy from the nodules was demanded from interventional radiology department with suspicious of metastases. She had no history of smoking but recurrent epistaxis since childhood. Her family history did not reveal any kind of genetic disorder concerning vascular abnormalities or hemorrhagic states. Physical examination did not reveal neither signs of any telangiectasia in perioral regions, oral mucosae nor changes in skin anywhere of the body. Any neurologic or gastrointestinal complain or laboratory abnormality was noted. Before biopsy procedure, we reevaluated prior CT and we had felt suspicious about the lesions since few of them had continuity with curvilinear structures suggestive of vessels. Hence, we performed pulmonary CT angiography (CTA) (Asteion 4, Toshiba Medical Systems, Japan) for further characterization. Pulmonary CTA showed multiple homogenous enhancing mass-like lesions which had connectivity between branches of dilated pulmonary artery and vein branches. These lesions were considered to be pulmonary AVMs. The biggest one, measured 40 x 28 mm, located in the right lower lobe (Figure 1). Additionally, CTA demonstrated enlargement of hepatic artery and distal branches along with periphery of liver parenchyma and markedly heterogeneous hepatic enhancement pattern (Figure 2). After then, we performed gray-scale and color Doppler ultrasonography which revealed enlarged and tortuous hepatic artery and its branches continuing till the periphery of the liver. Spectral analyze demonstrated high velocity low resistance flow with increased flow volume (Figure 3). According to these imaging findings and the history of epistaxis, the diagnosis is "possible" HHT and therefore we refused to make biopsy which could cause fatal results such as massive hemoptysis or aspiration. Then the patient was directed to manage endovascular treatment. The patient underwent coil embolization for the biggest AVM in right lower lobe with an uneventful post procedural period since then. The patient refused to have genetic investigation neither for her nor her first degree relatives.
DISCUSSION
Hereditary hemorrhagic telangiectasia (HHT) is a hereditary rare disease which characterized by the presence of multiple telangiectases and arteriovenous malformations (AVMs) in characteristic locations that have direct connections between arteries and veins. The clinical diagnosis of HHT is based on the Curacao criteria which are 1) spontaneous and recurrent epistaxis, 2) mucocutaneous telangiectases located in lips, oral cavity, nose and fingers, 3) visceral AVMs located in the lung, brain, liver or gastrointestinal tract, and 4) family history (2). If the patient has three of these four criteria, the diagnosis of HHT is "definite"; two of them are considered "possible" or "suspected", and one or none of them are considered "unlikely" (3). Our patient had a diagnosis of possible HHT depending on history of epistaxis since childhood and visceral AVMs in lungs and liver. The patient refused to have genetic investigation neither for her nor her first degree relatives. Also, she did not have mucocutaneous telangiectasia. So, it was not possible for her to make diagnosis of "definite" HHT.
Recurrent and spontaneous epistaxis is the most common findings and approximately occurs in 95% of individuals. Affected individuals have mucocutaneous telangiectasias in face, oral cavity and hands within the first decade of life. The prevalence of telangiectasias are similar to epistaxis. Visceral AVMs can be found commonly in the liver, lung or brain. Cerebral AVMs are congenital and cause intracranial hemorrhage which can be first symptom in children (1).
80-90% of the patients who have pulmonary AVMs (PAVMs) have HHT. Conversely, pulmonary AVMs occur in 37-45% of patients with HHT (4). The patients may have single or multiple PAVMs. The incidence of single and multiple lesions are 42-74% and 8-20%, respectively. If the PAVM is single, the most common location is the left lower lobe. Multiple PAVMs commonly occur in lower lobes bilaterally (5). Although, PAVMs are an infrequent, they may be an important component of differential diagnosis list of pulmonary nodules. The first step of radiological diagnosis is chest plain film which demonstrates abnormality in about 98% of patients. The classic radiographic finding of PAVM is a well defined and commonly lobulated round or oval mass, ranging from 1 to 5 cm in diameter. Contrast enhanced CT is an another useful and widely available diagnostic tool in patients who have suspicious of PAVM and it also can depict vascular anatomy of PAVM clearly. Additionally, CT may be helpful for detecting coexisting lung diseases (6). Magnetic resonance imaging (MRI) has some limitations in the diagnosis of PAVMs. PAVMs have low signal intensity with signal void areas due to rapid blood flow and this appearance may be seen in calcified or cystic lesion. Hence, it is hard to differentiate PAVMs from these lesions by MRI (7). Although CT and MRI could be helpful in the diagnosis of PAVMs, the gold standard diagnostic method is still pulmonary angiography (PA) (6). PA not only detects the lesions, but also shows communications between the lesion and pulmonary vascularies that is important for embolotheraphy or surgical resection (9).
The frequency of hepatic AVMs in HHT is 31-41% of cases (10). Patients with hepatic involvement are usually asymptomatic. But, diagnosis of hepatic involvement in suspected patients is important because it may cause portal hypertension, congestive heart failure, portosystemic encephalopathy, cholangitis. Digital subtraction angiography (DSA) of the hepatic artery is accepted to be gold standard for evaluating liver in HHT. But, it is an invasive technique and the newer technologies in CT and MRI allow assessment of hepatic changes easily. The routine screening for liver involvement is not recommended except in patients who meet only 1-2 criteria for HHT and the presence of hepatic involvement could make a diagnosis of "definitive" HHT (10). In our patient, liver function tests revealed normal levels and hepatic involvement of the disease was thought to be asymptomatic as it was an incidental finding. But, we were almost sure that dyspnea which was the presentation symptom of the patient emerged from pulmonary AVMS, only since her echocardiography was within normal limits initially and dyspnea of the patient was regressed after the embolization of greatest pulmonary AVM.
Radiologic evaluation plays an important role in identifying visceral involvement in HHT. Especially, in the patients who have suspicious lung lesions, AVM should be kept in mind in order to avoid biopsy resulting catastrophic consequences. Also, imaging techniques particularly contrast enhanced CT and CTA provide prognostic evaluation and therapeutic approach for HHT. Interventional procedures for AVMs should be preserved for treatment options including embolization or surgical resection.
ACKNOWLEDGEMENTS
A part of this manuscript was presented in National Congress of Radiology which was held between on 7-11 November 2012 in Susesi Hotel, Antalya.
CONFLICT of INTEREST
None declared.
REFERENCES
- Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995;333:918-24.
- Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66-7.
- McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genetics IN Medicine 2011;13:607-16.
- Faughnan ME, Granton JT, Young LH. The pulmonary vascular complications of hereditary haemorrhagic telangiectasia. Eur Respir J 2009;33:1186-94.
- Khurshid I, Downie G H. Pulmonary arteriovenous malformation. Postgrad Med J 2002;78:191-7.
- Gossage JR, Kanj G. Pulmonary arteriovenous malformations: A state of the art review. Am J Respir Crit Care Med 1998;158:643-61.
- Remy J, Remy-Jardin M, Wattinne L, Deffontaines C. Pulmonary arteriovenous malformation: evaluation with CT of the chest before and after treatment. Radiology 1992;182:809-16.
- Silverman JM, Jubien PJ, Herkens RJ, Pelc NJ. Magnetic resonance imaging evaluation of pulmonary arteriovenous malformation. Chest 1994;106:1333-8.
- Lee DW, White RI, Egglin TK, Pollak JS, Fayad PB, Wirth JA, et al. Embolotherapy of large pulmonary arteriovenous malformations: long term results. Ann Thorac Surg 1997;64:930-9.
- Buscarini E, Plauchu H, Garcia-Tsao G, White Jr RI, Sabba C, Miller F, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int 2006;26:1040-6.
Yazışma Adresi (Address for Correspondence)
Dr. Bet?l KIZILDAĞ
Kahramanmaraş S?t?? İmam ?niversitesi Tıp Fak?ltesi,
Radyoloji Anabilim Dalı,
KAHRAMANMARAŞ - TURKEY
e-mail: dr.betulkizildag@gmail.com