Sea-blue histiyositoz akci?er tutulumu
Ersin G?naY1, Selma FIrat G?ven1, Zafer Akta?1, Tu?rul ??P?T1, Yetkin A?aÇkIran2, Hakan Ert?rk3
1 Atat?rk G???s Hastal?klar? ve G???s Cerrahisi E?itim ve Ara?t?rma Hastanesi, G???s Hastal?klar? Klini?i, Ankara,
2 Atat?rk G???s Hastal?klar? ve G???s Cerrahisi E?itim ve Ara?t?rma Hastanesi, Patoloji Klini?i, Ankara,
3 Atat?rk G???s Hastal?klar? ve G???s Cerrahisi E?itim ve Ara?t?rma Hastanesi, Radyoloji Klini?i, Ankara.
?ZET
Sea-blue histiyositoz akci?er tutulumu
Sea-blue histiyositoz Niemann-Pick hastal???n?n alt? tipinden biridir. ?ocukluk ?a??nda ba?layan hepatosplenomegali olmas?, n?rolojik tutulumun olmamas? ve sfingomiyelinaz aktivitesinin azalmas?yla karakterizedir. Akci?er tutulumu sea-blue histiyositozda g?r?len nadir bir klinik tutulumdur. Bu olgu sunumunda, 15 ya??ndayken sea-blue histiyositoz tan?s? konulan 39 ya??ndaki bir erkek hasta sunulmaktad?r. Hasta klini?imize prod?ktif ?ks?r?k, hemoptizi, ate? ve kilo kayb?yla ba?vurdu. Hastan?n semptomlar?nda antibiyotik tedavisine ra?men gerileme olmamas? ?zerine ileri ara?t?rma sonucunda sea-blue histiyositozun akci?er tutulumu oldu?u g?r?ld?. ?lgin? olarak, hastan?n yak?nmalar? tan?sal bronkoalveoler lavaj sonras? d?zeldi. Nadir g?r?lmesi nedeniyle hasta literat?r e?li?inde tart???ld?.
Anahtar Kelimeler: Niemann-Pick hastal???, pn?moni, sea-blue histiyositoz.
SUMMARY
Pulmonary involvement in sea-blue histiocytosis
Ersin G?naY1, Selma FIrat G?ven1, Zafer Akta?1, Tu?rul ??P?T1, Yetkin A?a?kIran2, Hakan Ert?rk3
1 Clinic of Chest Diseases, Ataturk Chest Diseases and Chest Surgery Training and Research Hospital,
Ankara, Turkey,
2 Clinic of Pathology, Ataturk Chest Diseases and Chest Surgery Training and Research Hospital,
Ankara, Turkey,
3 Clinic of Radiology, Ataturk Chest Diseases and Chest Surgery Training and Research Hospital,
Ankara, Turkey.
Sea-blue histiocytosis is one of the six types of Niemann-Pick disease. It is characterized by childhood onset of hepatosplenomegaly, lack of neurological involvement and diminished sphingomyelinase activity. Pulmonary system is rarely involved sea-blue histiocytosis. In this paper, we present a 39-years-old male who had previously diagnosed as sea-blue histiocytosis at the age of 15. He was admitted to our clinic due to productive cough, hemoptysis, fever and weight loss. His symptoms did not resolve with the antibiotic treatment and further investigations revealed pulmonary involvement of sea-blue histiocytosis. After diagnostic bronchoalveolar lavage, his symptomes were improved, interestingly. This rare entity was discussed with literature survey.
Key Words: Niemann-Pick disease, pneumonia, sea-blue histiocytosis.
Geli? Tarihi/Received: 23/11/2011 - Kabul Edili? Tarihi/Accepted: 08/01/2012
INTRODUCTION
Niemann-Pick disease (NPD) is a lipid storage disease with six subgroups (NPD type A to F). NPD type F, also named as sea-blue histiocytosis (SBH), is seen in adult population (1). Foamy macrophages are called as sea-blue histiocytes that accumulate in visceral organs such as liver, spleen and rarely other organs due to decreased activity of sphingomyelinase. On the contrary, central nervous system is preserved in all cases that is characteristic feature of the disease. Pulmonary involvement in SBH is rare and may be asymptomatic if the disease is only limited within pulmonary system. However, it may also cause mortality and morbidity if respiratory insufficiency develops. Here we report a patient with SBH and pulmonary involvement and review the pertinent literature.
Case Report
A 39-years-old male patient was admitted to our clinic with complaining of cough, purulent sputum, hemoptysis, dyspnea on exertion, fever and weight loss of 4 kg in a month. Previously, at the age of 15 when he was examined for stomach ache, hepatosplenomegaly was discovered and fine-needle aspiration biopsies of liver and bone marrow were performed. The biopsy specimens demonstrated foamy macrophages. Sphingomyelinase defficiency was also detected, hence he received the diagnosis of SBH. Family history was unremarkable. On physical examination, vital signs were within normal limits, rales were present on the lower half of the left hemithorax. Spleen was palpated about 14 cm and liver was palpated about 12 cm under the rib cage. Neurological examination did not reveal any positive findings.
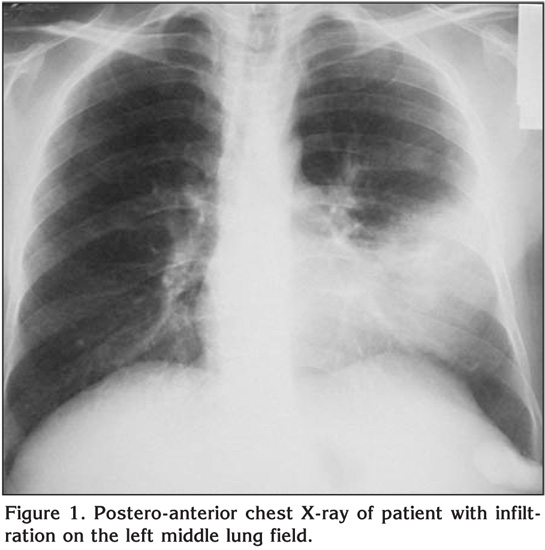
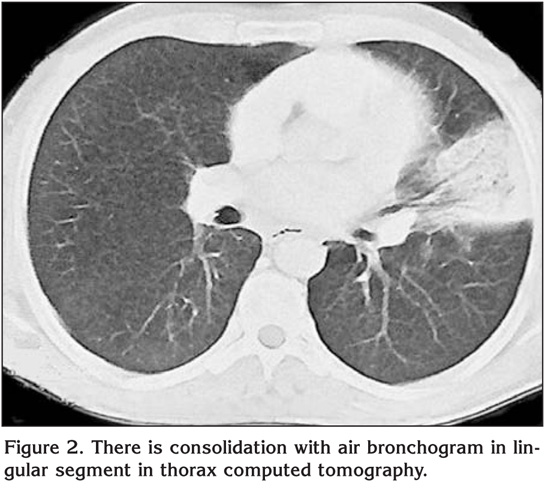
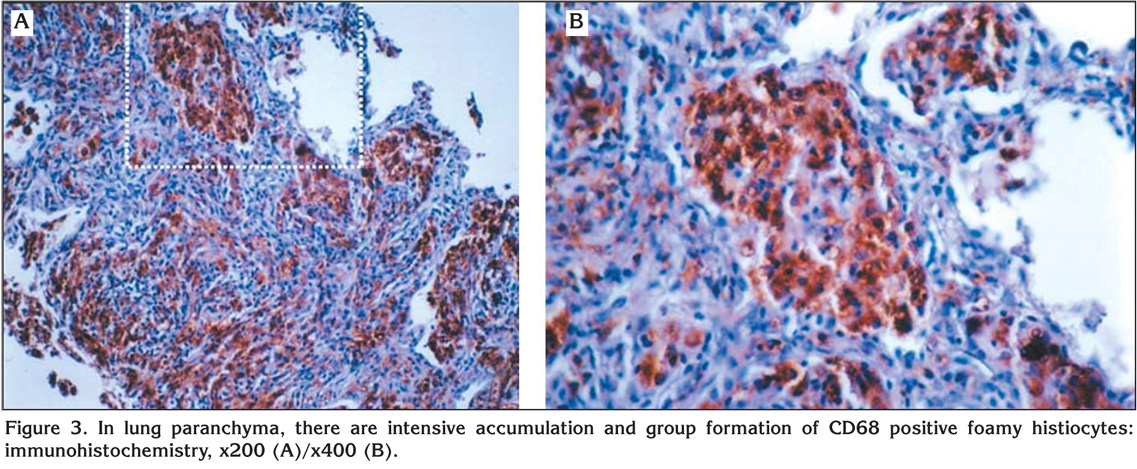
Laboratory findings were as below; erytrocyte sedimentation rate was 60 mm/hour and serum lipids were elevated [total serum cholesterol: 310 mg/dL (normal range: 140-240 mg/dL), high-density lipoprotein: 9 mg/dL (normal range: 30-85 mg/dL), low-density lipoprotein: 269 mg/dL, Very low-density lipoprotein: 33]. Pulmonary function tests revealed a restrictive pattern with FVC: 2.91 L (75% of predicted), FEV1: 2.15 L (65% of predicted) and FEV1/FVC: 74%. Carbon monoxide diffusion capacity was lower than normal; DLCO: 5.6 mmol/kPa.min (61% of predicted) and DLCO/VA: 1.2 mmol/kPa.min (74% of predicted). Arterial blood gas analysis in room air revealed pH: 7.38, PaO2: 40 mmHg, PaCO2: 42 mmHg. On the postero-anterior chest X-ray, consolidation was present in the lower zone of the left lung (Figure 1). Abdominal ultrasound revealed hepatosplenomegaly with craniocaudal length of liver and spleen 20 cm and 19 cm respectively. There was consolidation with air bronchogram at the lingula in thorax computed tomography (CT) sections (Figure 2). Three sputum samples were negative for acid-fast bacilli and tuberculosis skin test was negative. Lipid-poor diet was given to patient for hypercholesterolemia. Although non-specific antibiotic treatment of amoxicillin-clavulanic acid and clarithromycin was administered, no clinical or radiological improvement was achieved. For this reason, fiberoptic bronchoscopy was performed, which showed no endobronchial lesion. Bronchioloalveolar lavage (BAL) fluid was composed of 61% macrophages, 35% lymphocytes, and 4% neutrophiles without any cells with atypical features. After lavage procedure, an increase in oxygen saturation from 75.4% to 96.3% was detected. Cough and hemopytisis of the patient completely resolved in a few days. However, the consolidation seen on the radiographies persisted. Therefore, we performed CT guided tru-cut biopsy from consolidation area in lingular segment to exclude any malignancy. On microscopic examination of the biopsy specimen, lipid laden foamy histiocytes were seen with immunohistochemically expression of CD 68 covering the lung parenchyma (Figure 3). These findings were consistent with pulmonary involvement of SBH. Since pulmonary symptoms including hemoptysis were completely disappeared following BAL and declining of serum cholesterol levels was achieved with diet modification. No further treatment modality was planned. In his follow-up, the symptoms did not reoccur but the consolidation in the lingular segment persisted even after three years of diagnosis of pulmonary involvement.
Discussion
NPD consists of a group of congenital lipidoses autosomal recessively inherited in which sphingolipids accumulate in cells, particularly reticuloendothelial cells, throughout the body. Six types of NPD has been described. SBH, known as type F of NPD, is a rare type without neurological involvement. SBH is generally seen in adults (1,2,3). Sea-blue histiocyte is a macrophage that contains varying numbers of cytoplasmic granules imparting a distinct blue color on Wright's-Giemsa stain (4). In 1970, Silverstein et al. have described the syndrome of sea-blue histiocytes, characterized the presence of large foamy macrophagic cells in the bone marrow, spleen, and sometimes liver and lungs (5,6). SBH can be seen in different diseases of which hematological involvement such as chronic myeloid leukemia, idiopathic thrombocytopenic purpura, and myelodysplastic syndromes and metabolic involvement such as NPD and Gaucher diseases (6,7,8,9). SBH is differentiated from these diseases with sphingomyelinase defficiency and clinical appearance (7). Symptoms of Sea-blue histiocytes differ depending on the organs involved. NPD type A and C are seen in childhood and have poor prognosis. In SBH, hepatosplenomegaly is common. Other organ involvements are rare, as well neurological involvement is very rare (1). Our patient was 39-years-old. Although, he was first diagnosed when he was 15-years-old, he did not have any other complaints except for stomach ache for the following two decades. As expected, he had no neurological symptoms either.
Pulmonary pathologies are rarely encountered in NPD (8,10). In literature, there are only few cases about pulmonary involvement of SBH. In NPD, at the beginning, lipid-laden macrophages accumulate in lower lobes of lung fields which can be seen as diffuse linear or nodular infiltration in chest X-ray. Subsequently, upper lung fields may also be involved. In thorax tomography, interlobular septal thickening, multiple pulmonary nodules and ground-glass appearances can be seen secondary to partial accumulation of alveoli with foamy histiocytes. Less commonly, lenfadenopathies, pleural involvement and cavitation may be present (8,10). In our case, CT revealed nodular interstitial thickening, ground-glass infiltration with air bronchogram and bronchiectasis. In NPD type B, although pulmonary involvement can be asymptomatic, it may be the main cause of mortality and morbidity if it leads to respiratory insufficiency (10).
There is no known effective treatment of pulmonary involvement of NPD. A lot of therapeutic modalities have been applied for palliation. If respiratory failure with hypoxia was present, nasal oxygen supplement can be provided (1). If bilateral lung involvement is present, patient can be succesfully treated with whole lung lavage (10). After application of diagnostic BAL of lingular segment, interestingly, oxygen saturation of our patient increased, but, infiltration on chest X-ray was not be resolved. We did not need to apply whole lung lavage because of limited involvement of lingular segment. Since lipid poor diet may be effective for hiperlipidemia in NPD we also modified the patient' diet and achieved improvement in serum lipid levels (1).
There are some additional treatment alternatives, which are not routinely used. One of them is stem cell transplantation which was claimed effective for NPD (11,12). Also, bone marrow transplantation have shown improvement in lung infiltrations and provided regression of hepatomegaly (13). Additionally, amniotic epithelial cell implantation can be effective for sphingomyelinase deficiency (14,15). Enzyme replacement therapy and gene therapy are promising but further studies are required.
In conclusion, lysosomal storage diseases are rare in the population and pulmonary involvement is not a common feature of this group of diseases. Nevertheless, lipidosis with lung involvement should be considered in differential diagnosis of the patients with persistent pulmonary infiltrates, particularly of those with a pertinent personal or family history.
CONFLICT of INTEREST
None declared.
REFERENCES
- Schwartz RA, 30 April 2008, Niemann-Pick Disease. Available from: http://www.emedicine.com/derm/topic699.htm. Accessed date: 17.11.2008.
- Girisgen ?, Karakas T, Yaris N, Okten A. Niemann-Pick Type B. Turkiye Klinikleri. J Pediatr 2007; 16: 137-9.
- G???? S, G??men A, Ko?ak N, Kiper N, K???kali T, Y?ce A, et al. Lipidosis with sea-blue histiocytes. Report of two siblings with lung involvement. Turk J Pediatr 1994; 36: 139-44. [?zet]
- Landas S, Foucar K, Sando GN, Ellefson R, Hamilton HE. Adult Niemann-Pick disease masquerading as sea blue histiocyte syndrome: report of a case confirmed by lipid analysis and enzyme assays. Am J Hematol 1985; 20: 391-400. [?zet]
- Silverstein MN, Ellefson RD, Ahern EJ. The syndrome of the sea-blue histiocyte. N Engl J Med 1970; 282: 1-4. [?zet]
- Candoni A, Grimaz S, Doretto P, Fanin R, Falcomer F, Bembi B. Sea-blue histiocytosis secondary to Niemann-Pick disease type B: a case report. Ann Hematol 2001; 80: 620-2. [?zet]
- Schneider EL, Pentchev PG, Hibbert SR, Sawitsky A, Brady RO. A new form of Niemann-Pick disease characterised by temperature-labile sphingomyelinase. J Med Genet 1978; 15: 370-4. [?zet] [PDF]
- Minai OA, Sullivan EJ, Stoller JK. Pulmonary involvement in Niemann-Pick disease: case report and literature review. Resp Med 2000; 94: 1241-51. [?zet]
- Suzuki O, Abe M. Secondary sea-blue histiocytosis derived from Niemann-Pick Disease. J Clin Exp Hematopathol 2007; 47: 19-21. [?zet]
- Nicholson AG, Wells AU, Hooper J, Hansell DM, Kelleher A, Morgan C. Successful treatment of endogenous lipoid pneumonia due to Niemann-Pick Type B disease with whole-lung lavage. Am J Respir Crit Care Med 2002; 165: 128-31. [?zet] [Tam Metin] [PDF]
- Schuchman EH. Hematopoetic stem cell gene therapy for Niemann-Pick disease and other lysosomal storage diseases. Chem Phys Lipids 1999; 102: 179-88. [?zet]
- Winchester B, Vellodi A, Young E. The molecular basis of lysosomal storage disease and their treatment. Biochem Soc Trans 2000; 28: 150-4. [?zet]
-
Vellodi A, Hobbs JR, O'Donnell NM, Coulter BS, Hugh-Jones K. Treatment of Niemann-Pick
disease type B by allogeneic bone marrow transplantation. Br Med J (Clin Res
Ed) 1987; 295: 1375-6.
[?zet] [PDF] - Bembi B, Comelli M, Scaggiante B, Pineschi A, Rapelli S, Gornati R, et al. Treatment of sphingomyelinase deficiency by repeated implantations of amniotic epithelial cells. Am J Med Genet 1992; 44: 527-33. [?zet]
-
Scaggiante B, Pineschi A, Sustersich M, Andolina M, Agosti E, Romeo D. Successful therapy
of Niemann-Pick disease by implantation of human amniotic membrane.
Transplantation 1987; 44: 59-61.
[?zet]
Yaz??ma Adresi (Address for Correspondence):
Dr. Ersin G?nay,
I?d?r Devlet Hastanesi,
2. G???s Hastal?klar? Poliklini?i,
I?DIR - TURKEY
e-mail: ersingunay@gmail.com