?nterstisyel akci?er hastal???n?n nadir bir nedeni: Hermansky-Pudlak sendromu
Ayd?n
??LEDA?1, Burcu C?R?T KO?ER1, Nurdan K?KT?RK2,
Ak?n KAYA1, G?khan ?EL?K1,
Numan NUMANO?LU1
1 Ankara ?niversitesi T?p Fak?ltesi, G???s Hastal?klar? Anabilim Dal?, Ankara,
2 Gazi ?niversitesi T?p Fak?ltesi, G???s Hastal?klar? Anabilim Dal?, Ankara.
?ZET
?nterstisyel akci?er hastal???n?n nadir bir nedeni: Hermansky-Pudlak sendromu
Hermansky-Pudlak sendromu kanama diyatezi, ok?lok?tan?z albinizm ve dokularda lizozomal "ceroid" lipofuskin pigment depolanmas? ile karakterize nadir bir hastal?kt?r. Hastal??a pulmoner fibrozis de e?lik edebilmektedir. K?rk sekiz ya??nda Hermansky-Pudlak sendromu tan?s? ile izlenen erkek hasta, nefes darl??? ?ikayeti nedeniyle ba?vurdu. Toraks bilgisayarl? tomografide her iki akci?er alt lob bazal segmentlerde daha belirgin olmak ?zere yayg?n interlob?ler septal kal?nla?malar saptand?. Olguda pirfenidon tedavisi planlanmas?na ra?men, klinik bozulma geli?ti ve hasta solunum yetmezli?i nedeniyle kaybedildi. Sonu? olarak bu yaz?da, olduk?a nadir ve mortal bir hastal?k olan Hermansky-Pudlak sendromunun akci?er tutulumuna ba?l? pulmoner fibrozisli bir hasta sunulmaktad?r.
Anahtar Kelimeler: Hermansky-Pudlak sendromu, interstisyel akci?er hastal???.
SUMMARY
A rare cause of interstitial lung disease: Hermansky-Pudlak syndrome
Ayd?n
??LEDA?1, Burcu C?R?T KO?ER1, Nurdan K?KT?RK2,
Ak?n KAYA1, G?khan ?EL?K1,
Numan NUMANO?LU1
1 Department of Chest Diseases, Faculty of Medicine, Ankara University, Ankara, Turkey,
2 Department of Chest Diseases, Faculty of Medicine, Gazi University, Ankara, Turkey.
Hermansky-Pudlak syndrome is a rare disease characterized by bleeding diathesis, oculocutaneous albinism and lysosomal ceroid lipofuscin pigment deposits. Pulmonary fibrosis may also accompany with the disease. A 48-year-old male patient with a diagnosis of? Hermansky-Pudlak syndrome admitted with dyspnea. A thorax computed tomography revealed bilateral diffuse interlobular septal thickness which was more prominent in the basal segments of lower lobes. Although pirfenidone therapy was planned, clinical deteroriation developed and patient died because of respiratory failure. In conclusion; this report describes a patient with pulmonary fibrosis caused by lung involvement of Hermansky-Pudlak syndrome which is an extremely rare and mortal disease.
Key Words: Hermansky-Pudlak syndrome, interstitial lung disease.
Hermansky-Pudlak syndrome (HPS), first described in 1959, is characterized by a classic triad of oculocutaneous albinism, a platelet storage pool deficiency and lysosomal accumulation of ceroid lipofuscin (1). Ceroid lipofuscin, in particular, contributes much to the morbitidy associated with the disease, as ceroid deposition affects many organ systems and is especially problematic in the lungs where it is often associated with pulmonary fibrosis (2).
HPS is an autosomal recessive disease. Although worldwide it is extremely rare, HPS is most common in northwest Puerto Rico, where its frequency is 1 in 1800 (3). Inheritance of one of the eight known HPS genes produces one of eight described subtypes of HPS, HPS1 through HPS8, with HPS1 and HPS4 showing the greatest degree of pulmonary involvement (4,5).
CASE REPORT
A 48-year-old male patient who was diagnosed as HPS according to the history of repeated gastrointestinal tract bleeding, typical fenotypic findings, bleeding diathesis caused by impaired thrombocyte functions which was confirmed by electron microscopy of platelets demonstrating absent dense bodies, was admitted to our clinic with five months history of gradually increasing dyspnea. The patient was otherwise asymptomatic. He had 10 pack-year of smoking history and no occupational exposure to known organic or inorganic agents. He was receiving proton pump inhibitor for two years and taking not any other medication. There was no family history of HPS.
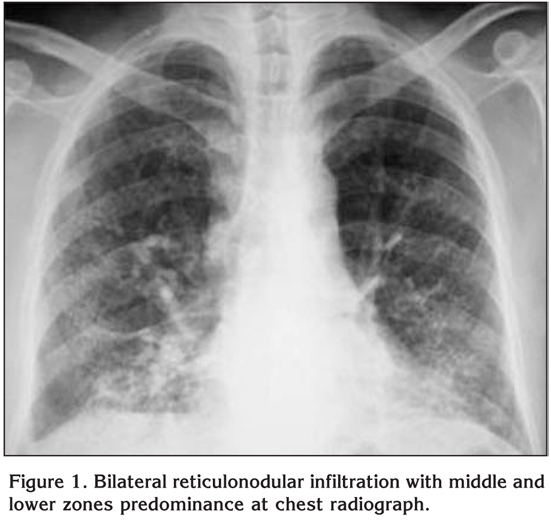
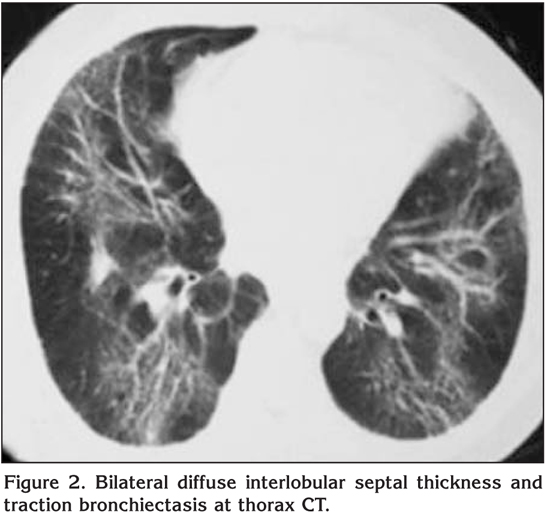
On physical examination oculocutaneous albinism, strabismus, nystagmus, clubbing and bilateral inspiratory rales were detected. The temperature was normal, the blood pressure 100/60 mmHg, the pulse 112 beats per minute and the respiratory rate 28 breaths per minute. Laboratory studies revealed a normal complete blood count and serum biochemistry. The prothrombin time, partial-thromboplastin time and international normalized ratio were in normal range, however, the bleeding time was prolonged (14 minutes; normal: 2-9 minutes). Chest radiograph showed bilateral reticulonodular infiltration with middle and lower zones predominance (Figure 1). A thorax computed tomography (CT) revealed bilateral diffuse interlobular septal thickness and traction bronchiectasis which were more prominent in the basal segments of lower lobes and also a honeycomb pattern (Figure 2). The oxygen saturation was 66% while the patient was breathing ambient air and arterial blood gas analyses revealed severe hypoxemia with a PaO2 value of 36.7 mmHg. Accordingly, oxygen therapy was initiated. Since supplemental oxygen by nasal cannula failed to increase the oxygen saturation above 90%, oxygen was given by a face mask with reservoir bag. During follow-up, although pirfenidone therapy was planned, clinical deteroriation developed and the patient died at the 14th day of hospitalization because of the respiratory failure.
DISCUSSION
HPS is a type of oculocutaneous albinism associated with a bleeding diathesis and pulmonary fibrosis. Although its frequency is 1 in 1800 in northwest Puerto Rico, worldwide it is an extremely rare disorder with a prevalence of 1 in 500.000 to 1 in 1.000.000 (6,7).
The syndrome comprises eight known autosomal recessive disorders (HPS1 to HPS8) (5). HPS1 is the most common subtype. The genes are lacking in the HPS code for proteins that function in organelle biogenesis and transport of lysosomes and lysosome-related organelles.
The findings in HPS are related to a dysfunction in biosenthesis of membrane-bound organelles: melanosomes in oculocutaneous albinism, platelet dense bodies in bleeding dysfunction and lysosomes in ceroid lipofuscin deposition. The dysfunction of melanosomes causes oculocutaneous albinism including hypopigmentation of the skin, hair and reduced iris and retinal pigment. The other ocular manifestations include visual impairment, horizontal nystagmus and strabismus (8). The bleeding diathesis may result in easy bruising, epistaxis, menorhagia, postpartum hemorrhage, gingival bleeding, colonic bleeding and prolonged bleeding after dental extraction or surgery. In presented case, the bleeding diathesis was emerged as intermittent gastrointestinal system bleeding and epistaxis.
Pulmonary fibrosis is the most serious complication and main reason of death of patients with HPS (9,10). It usually presents in the 3rd or 4th decades of life and accounts for premature death in 50% of HPS patients, generally by the 5th decade. The incidence and severity of fibrosis is greatest in patients with HPS1 and HPS4.
Pathogenesis underlying the pulmonary fibrosis is unclear. It has been suggested that continual deposition of ceroid lipofuscin distrupts type II alveolar cells and leads to chronic inflammation and progressive fibrosis (2). The dysfunction of lamellar bodies of type II alveolar cells is probably also a contributing factor (11).
The radiologic appearance of pulmonary fibrosis in HPS is comparable to that of idiopathic pulmonary fibrosis and the predominant radiologic pattern is interstitial patterns or infiltrates that usually involve both lungs symmetrically as in our case.
The general outcome for patients with HPS varies depend on the disease severity. The ocular component of HPS results legal blindness. The skin changes may predispose to cancer such as basal cell and squamous cell cancer and sunscreen and avoidance of sunlight are important measures for decreasing the risk. Although the bleeding diathesis is a major concern during surgery or dental extraction, it rarely results in fatal complications. Desmopressin may be used prophylactically. Recombinated activated factor VII (VIIa) has also been reported in successfully shortening the bleeding time. Avoidance of aspirin products is essential.
As mentioned above, pulmonary fibrosis is the main cause of mortality in HPS. Lung transplantation is the only known treatment for pulmonary fibrosis and it has been performed successfully in only one patient to date (12). Steroid therapy is not an effective treatment (2). Pirfenidone, a pyridine molecule with anti-inflammatory and antifibrotic activites is a promising agent in the treatment and it has been shown to slow the progression of fibrosis but only in patients who have significant residual lung function (9). In our case, although pirfenidone therapy was planned, due to absence of this agent in our country it could not be given and the patient died at the 14th day of hospitalization.
In conclusion, herein we presented a case with pulmonary fibrosis caused by the lung involvement of HPS which is an extremely rare and mortal disease.
CONFLICT of INTEREST
None declared.
REFERENCES
- Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigment reticular cells in the bone marrow: report of two cases with histochemical studies. Blood 1959; 14: 162-9.
- Pierson DM, Ionescu D, Qing G, et al. Pulmonary fibrosis in Hermansky-Pudlak syndrome. Respiration 2006; 73: 382-95. [?zet]
- Witkop CJ, Babcock MN, Rao GHR, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Assoc Med PR 1990; 82: 333-9. [?zet]
- Santiago Borrero PJ, Rodriguez-Perez Y, Renta JY, et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky-Pudlak syndrome type 1 and 3 mutations in Puerto Rico. J Invest Dermatol 2006; 126: 85-90. [?zet] [Tam Metin] [PDF]
- Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res 2006; 19: 19-42. [?zet]
- Izquierdo NJ, Royuela MA, Maumenee IH. Possible origins of the gene of Hermansky-Pudlak in Puerto Rico. PR Health Sci J 1993; 12: 147-8. [?zet]
- Poddar RK, Coley S, Pavord S. Hermansky-Pudlak syndrome in a pregnant patient. Br J Anaesth 2004; 93: 740-2. [?zet] [Tam Metin] [PDF]
- Tager AM, Sharma A, Mark EJ. A 27-year old man with progressive dyspnea. N Engl J Med 2009; 361: 1585-93.
- Gahl WA, Brantly M, Troendle J, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab 2002; 76: 234-42. [?zet]
- Harman KR, Witkop JC, White JG, et al. Pathogenesis of pulmonary fibrosis: platelet-derived growth factor precedes structural alterations in the Hermansky-Pudlak syndrome. J Lab Clin Med 1994; 123: 617-27. [?zet]
- Hurford MT, Sebastiano C. Hermansky-Pudlak syndrome: report of a case and review of the literature. Int J Clin Exp Pathol 2008; 1: 550-4. [?zet] [Tam Metin] [PDF]
- Lederer DJ, Kawut SM, Sonett JR, et al. Successful bilateral lung transplantation for pulmonary fibrosis associated with the Hermansky-Pudlak syndrome. J Heart Lung Transplant 2005; 24: 1697-9. [?zet]
Yaz??ma Adresi (Address for Correspondence):
Dr. Ayd?n ??LEDA?,
Ankara ?niversitesi T?p Fak?ltesi,
G???s Hastal?klar? Anabilim Dal?,
06100, Cebeci, ANKARA - TURKEY
e-mail: aciledag@yahoo.com